This page highlights several of my research projects. For a complete list of my publications, see my publications page.
Research
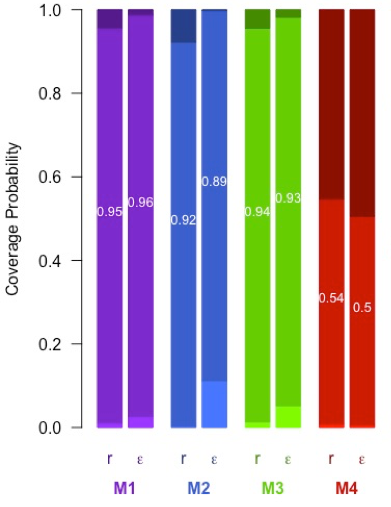
Coverage probability of the net diversification rate r and the turnover rate ε of 95% credible interval for M1 (Birth-Death process under bernoulli sampling with integrated sampling fraction), M2 (Birth-Death process under k-sampling), M3 (Birth-Death process under bernoulli sampling), M4 (Birth-Death process where the phylogeny is assumed complete).
Model development for analyzing the diversification of clades of unknown diversity
Institute of Biology of ENS, 2018 - 2022
The vast majority of the microbial diversity is still unknown. Despite, technical efforts in term of sequencing and model development, it is still challenging to estimate accurately the number of microbial species we are missing. When studying the diversification of a clade using a phylogeny, this estimate of global diversity is required. To overcome this limitation, we propose simple diversification models that do not require to estimate the global diversity of the clade of interest. All the models proposed are based on the birth-death process and assume that the diversification rates of the considered clade are constant through time and across lineages. The models either use the information on the number of sampled species in the phylogeny (called k-sampling model since we condition the process on k: the number of sampled species) or integrate on the whole possible sampling fraction range uniformly (from 0: no species sampled to 1: fully sampled phylogeny) assuming that each species has the same probability to be sampled (Bernoulli sampling scheme). The inference of the diversification rates can be done on a phylogeny or a set of phylogenies assuming common or distinct rates. A variant of those models where a specific sampling fraction distribution is assumed (Beta distribution) is also proposed. The inference is done using Bayesian inference and the models were tested on simulated and empirical dataset. The inference is fast (using closure for likelihood calculation) and as much accurate as we can get (dichotomic search to overcome some numerical approximation for some large phylogenies with low probability). The models are designed for simple diversification analysis with assumptions on rate homogeneity through time and across lineages while alleviating the need to estimate global diversity. It is particularly appropriate for diversification analysis on large microbial clades but also other largely under-sampled clade with unknown global diversity such as coleopterans.
Mentors: DR. Hélène Morlon, Pr. Amaury Lambert
Presented at 2019 Mathematical and Computational Evolutionary Biology (MCEB) at Porquerolles, FR and at 6th Young Natural History Scientists’ Meeting at MNHN, Paris, FR.
Publications: In preparation. Draft available upon demand.
Code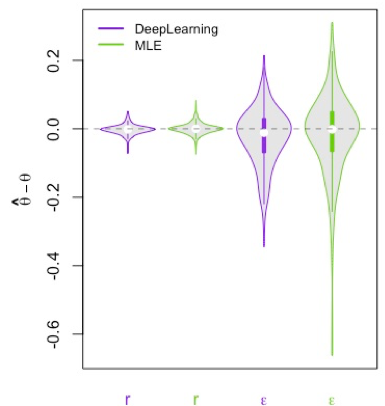
Bias distribution of the net diversification rate r and the turnover rate ε estimated using deep neural network (CNN) or maximum likelihood.
Inference technique for fast and flexible, likelihood free diversification analyses
Institute of Biology of ENS, 2018 - 2022
Diversification inference are currently based on likelihood calculations. Depending on the diversification model and the size of the dataset in hand the calculations of the likelihood and thus the inference can be time and computer power consuming. These limitations are becoming more and more prevalent due to the increasing availability of bigger dataset and the need to complexify models. This is preventing the analysis of large phylogenies using complex and more realistic diversification models. One way to overcome these limitations is to use a likelihood free inference machinery such as deep learning. To do so, we trained deep neural networks on simulated phylogenies assuming a diversification model. Several network architectures were tested using different types of input data (CNN: full tree representation, FFNN: summary statistics). The performance of the deep learning inference is comparable to the maximum likelihood estimation while being several order of magnitudes faster once the network is trained. This approach is faster but also more flexible since no likelihood of the model needs to be developed for the inference. Only the simulation of the process needs to be developed for the network to learn the relationship between the input data and the parameters of interest.
Mentors: DR. Hélène Morlon
Collaborators: Jakub Voznica
Will be prensented at Evolution Conference in June 2022 at Cleveland, US.
Publications: In preparation. Draft available upon demand.
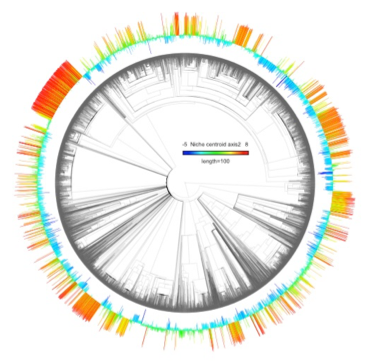
Phylogenetic distribution of the ecological niche dimension caracterised by nutrients. The warm and cold colors represent respectively nutrient rich and poor niches.
Diatoms diversification in light of their ecological niche space
Institute of Biology of ENS, 2018 - 2022
Diatoms are responsible for the majority of carbon fixation in the ocean. They are globally distributed and they particularly thrive at high latitude - a counter example of the common latitudinal diversity gradient. Diatoms have also large heterogeneity in their diversification dynamic. To understand this phenomena, we are characterizing diatoms ecological niche strategy (niche width, singularity…) and investigating the relationship between their strategy and their propensity to diversify.
Mentors: DR. Hélène Morlon, DR. Chris Bowler
Collaborators: Richard Dorrell
Presented at Virtual Evolution Conference and at 1rst Congrès des Jeunes Chercheurs du Muséum at MNHN, Paris, FR. Publications: In preparation.